INTRODUCCIÓN
La historia natural de la enfermedad de Paget fue descrita por primera vez por Sir James Paget en 1877 en su artículo titulado Osteítis deformante. La enfermedad de Paget es un desorden metabólico focal del hueso que generalmente aparece en personas mayores con una tasa aumentada de remodelación del hueso, que resulta en un aumento de volumen del hueso, puede ser de aparición focal o multifocal, generalmente afecta la Pelvis, parte superior de fémur, cráneo, columna vertebral, humero y tibia.
La mayoría de estas lesiones son asintomáticas y muchas de ellas se encuentran como hallazgo incidental en estudios de imágenes o como una elevación de la fosfatasa alcalina en exámenes de laboratorio. Las que son sintomáticas (5% de los pacientes que asisten a consulta médica), se manifiestan por dolor local, deformidad o compresión por crecimiento del hueso. El hueso neoformado es mucho más débil y permite la aparición de fracturas patológicas. Algunos casos han reportado eritema y aumento de la temperatura en el área del cráneo afectada, con aumento de vascularización de la lesión con la descripción de la presencia de soplos a la auscultación de la lesión. Se ha registrado una frecuencia del 1,3 al 9% de pacientes con edad avanzada y se comienza a observar a partir de los 55 años con leve predominio masculino, más frecuente en Europa, en Inglaterra se ha descrito una prevalencia de hasta 8,3%, rara en Escandinavia y Asia. En Estados Unidos se ha encontrado una prevalencia de 1-2%, siendo la segunda enfermedad del hueso después de la osteoporosis.
Los cambios radiológicos que se ven en la enfermedad de Paget son caracterizados por la destrucción del hueso, seguido por reparación del mismo. Estas dos fases pueden estar presentes en un mismo caso con expansión del hueso. La fase de destrucción se ve más frecuentemente en el cráneo y se le denomina osteoporosis circunscrita. Mientras que en la etapa de reparación el hueso se ve esclerótico, expandido y con gruesas trabéculas. En el cráneo los cambios suceden al inicio en la tabla externa y luego afecta ambas tablas craneales y se pierde la diferenciación entre ellas, llegando a aumentar el grosor hasta 5 veces su tamaño normal. Las lesiones osteoliticas pueden ser la única manifestación de la enfermedad inicial mientras que la radiodensidad y la trabeculación son características de un proceso avanzado. Se ha descrito afectación de la base del cráneo con Platibasia, impresión basilar que pueden producir síntomas piramidales por compresión del tallo cerebral, crecimiento óseo que atrapa a los nervios que pasan por los forámenes produciendo hipoacusia y atrofia óptica.
Desde el punto de vista del laboratorio se encuentran cifras elevadas de fosfatasa alcalina con valores séricos de calcio y fosforo dentro del rango normal. Otros marcadores de recambio óseo, reabsorción y formación ósea se han utilizado, como son la fosfatasa alcalina especifica de hueso, osteocalcina, procolágeno tipo I, C-propéptido y procolágeno tipo N-propéptido (formación) y marcadores urinarios de reabsorción óseas como la deoxipirridiolina y el N-telopeptido corregido con creatinina. Estos valores se correlacionan con los niveles de fosfatasa alcalina y aparentemente no definen mejor la enfermedad o la respuesta al tratamiento. Raramente se ha descrito transformación en osteosarcoma de la enfermedad de Paget, se presenta en el 1% de los casos . En pacientes con historia familiar de Paget, existe la tendencia a la aparición de lesiones en edades tempranas, más sitios con afectación ósea, acentuación de la deformidad y muchas más fracturas .
El hecho que existan las 2 modalidades de presentación esporádica y familiar y que existan áreas geográficas delimitadas con mayor incidencia, hace pensar en la posibilidad de factores ambientales o genéticos involucrados en la patología. En estudios genéticos se han encontrado mutaciones genéticas en un porcentaje de los pacientes, hoy en día se sabe que es un desorden autosómico dominante con penetración variable, los factores ambientales que modulan el fenotipo no han sido aislados todavía.
El tratamiento actual es con la nueva generación de bifosfonatos que ayudan al control de la enfermedad.
CASO CLÍNICO
Paciente femenino de 51 años de edad, con antecedente de hipertensión arterial, quien presento cefalea biparietal irradiada a región occipital, progresiva, intensa, atenuada con Ketorolac y exacerbada con la actividad física, concomitantemente fotofobia. Valorada por medicina interna quien hospitaliza con los diagnósticos de 1. Gammapatía monoclonal mieloma múltiple; 2. Nódulo pulmonar; 3. Hipertensión arterial; 4. Tabaquismo. Paciente presentaba como antecedentes personales hipertensión arterial desde hace 5 años, en tratamiento con Olmesartan, Midoxomil 20 mg/día, Asma bronquial en la infancia, histerectomía por miomatosis uterina que ameritó transfusión de hemoderivados. Exeresis de lipoma cervical y abdominal en el 2008 y como antecedentes familiares madre viva 85 años hipertensa con demencia senil, padre fallecido por cáncer de pulmón.
Al examen físico paciente presentaba dolor a la palpación en región parietal derecha, adenopatías latero cervicales bilaterales de 1 x 2 aproximadamente, no dolorosas, móviles, no adheridas a planos, resto del examen físico sin alteraciones. Dentro de los paraclínicos hematológicos la fosfatasa alcalina se encontraba elevada (ver Tabla 1). Estudio de rayos X de cráneo en proyección lateral mostraba múltiples imágenes osteoliticas, con engrosamiento del espesor del hueso (Figura 1), estudio de rayos X de torax, humero y femur se encontraban sin alteraciones (Figura 2, 3 y 4).
Tabla 1. Paraclínicos
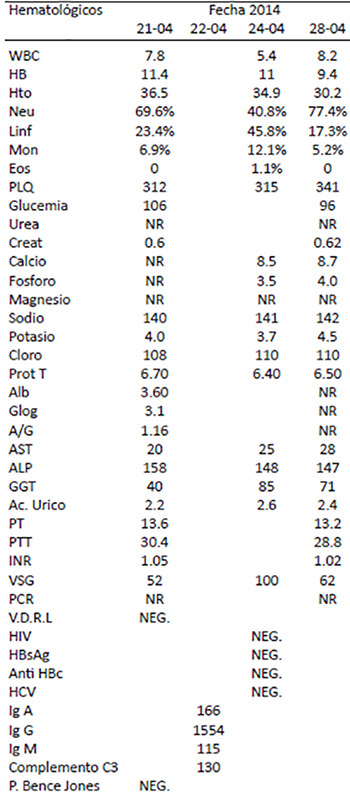
Figura 1. Imágenes osteológicas con áreas de mayor densidad que no respeta la sutura coronal, se aprecia espesor aumentado del grosor del hueso parietal con radiopacidad de la cortical interna y radiolucidez de la cortical externa.
Figura 2. Rayos X de torax, sin alteraciones.
Figura 3. Rayos X de húmero, sin alteraciones.
Figura 4. Rayos X de femur sin alteraciones.
Tomografía axial computarizada evidenciaba en ventanea ósea engrosamiento diploico de los huesos frontales y parietales a predominio del lado derecho, asi como imágenes hipodensas, irregulares en los huesos frontal y parietal, a predominio parietal derecho y mientras que ventana parenquimatosa se mostraba sin alteraciones (Figura 5). RMN cerebral con contraste evidencio imagen heterogénea en T1 y T2 que capta contraste en región ósea fronto-parieto-occipital bilateral a predominio derecho, resto sin alteraciones (Figura 6). Es llevada a procedimiento quirúrgico donde se realiza craniectomía parietal derecha evidenciándose aumento de la vascularización ósea y osteolisis acompañada de fragilidad.
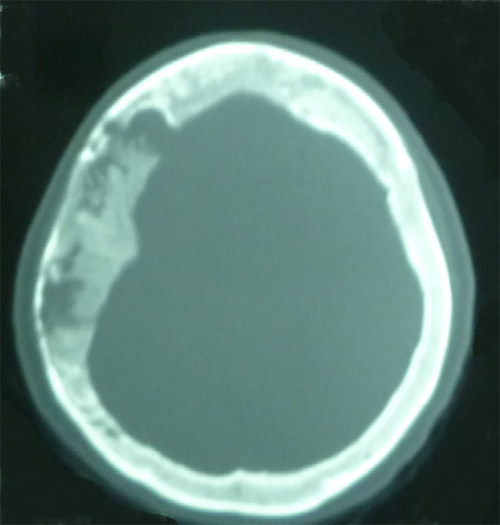
Figura 5. Tomografía axial computarizada en ventanea ósea que muestra engrosamiento diploico en huesos frontales y parietales a predominio derecho, imágenes hipodensa, irregulares en huesos frontales y parietales a predominio parietal derecho.
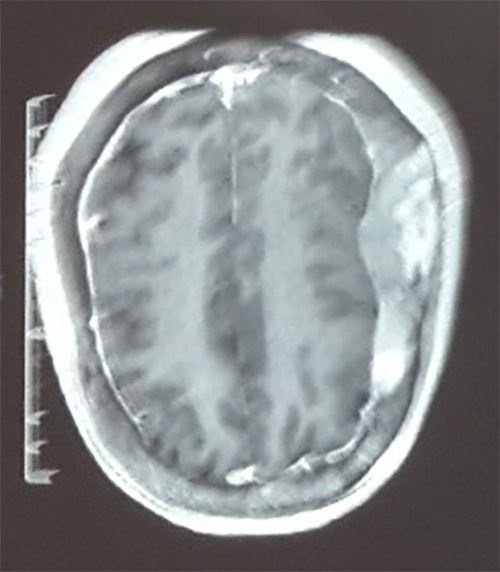
Figura 6. RMN cerebral con contraste donde se evidencia imagen heterogénea que capta contraste en región ósea fronto-parieto-occipital bilateral a predominio derecho.
Los estudios histopatológicos reportaron enfermedad de Paget, no observándose infiltración por lesión linfoproliferativa ni encontramos criterios para el diagnostico de otro tipo de neoplasia (Figura 7, 8 y 9). La inmunohistoquímica del tejido óseo osteoesclerótico fue negativa para el diagnóstico de neoplasia de células plasmáticas.
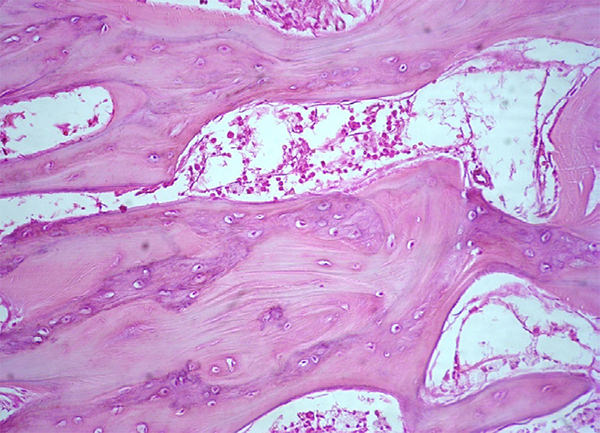
Figura 7. Hiperostosis, trabéculas óseas aumentadas de espesor, presencia de líneas de cemento prominentes.
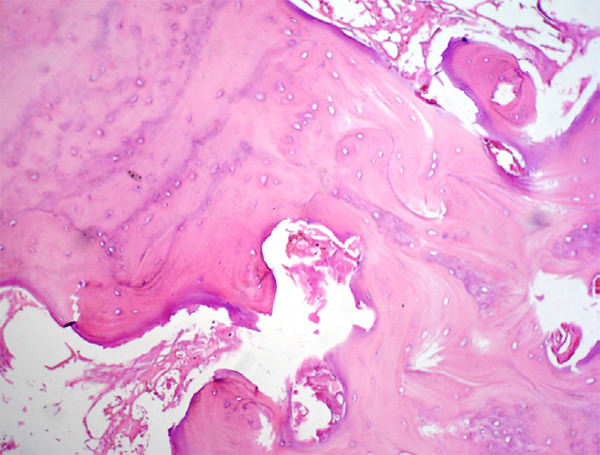
Figura 8. Hiperostosis, trabéculas óseas aumentadas de espesor, presencia de líneas de cemento prominentes. La médula ósea no muestra infiltración por tumor.
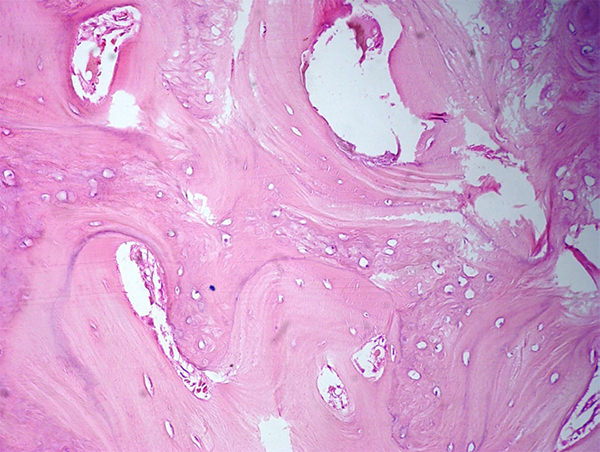
Figura 9. Trabéculas óseas aumentadas de espesor con líneas de cemento prominentes. Los canales de Havers libres de infiltración por neoplasia.
DISCUSIÓN
La enfermedad de Paget es un trastorno de remodelación ósea crónico, no inflamatorio, que afecta áreas no contiguas del esqueleto, es la segunda enfermedad ósea más frecuente después de la osteoporosis. Su prevalencia muestra una amplia variación geográfica, común en Europa occidental, América y Australia, pero rara en Asia y África. Sin embargo, estudios recientes han informado una reducción inexplicada en la prevalencia y la gravedad de la enfermedad. Esta enfermedad es relativamente común en las personas mayores, se presenta en aproximadamente el 3-4% de la población mayor de 50 años con una ligera predilección del género masculino.
En la actualidad se considera que factores tanto genéticos como ambientales están implicados en la patogénesis de la enfermedad, siendo los factores genéticos el componente más importante de la etiología, donde del 15-40% de los pacientes afectados tienen un familiar de primer grado con la enfermedad. Hasta el momento la enfermedad se comporta como autosómica dominante con penetración variable. Se han determinado 7 loci genéticos, el más documentado es la mutación P392L, asociada a la ubiquitina en el mapa genético PDB3 en locus 5q35 , esta mutación está presente en el 46% de los Paget familiar y en 16% de los de aparición esporádica. Se ha propuesto otro loci como 5q31 (locus PDB4) 2q36 y el 10p13 .
Histológicamente se observan osteoclastos de apariencia bizarra, multinucleados y en número aumentado, él hueso luce desorganizado con trabéculas de grosor variable rodeado de numerosos osteoblastos, la medula ósea es substituida por tejido estromal altamente vascularizado.
Clínicamente si la enfermedad no está avanzada el hueso puede ser normal, pero en casos avanzados la hipertrofia generada por la constate remodelación ósea puede exponer deformidades, con cambios térmicos importantes en la piel subyacente al fragmento de hueso patológico en el cráneo, es debido al aumento de la vascularización por el recambio óseo elevado. Aunque la mayoría de los pacientes son asintomáticos, los síntomas pueden resultar directamente de la implicación ósea (dolor de huesos, artritis secundaria y fracturas) o secundariamente a partir de la expansión de hueso que causa la compresión de tejido neural circundante y consecuente presencia de hipertensión endocraneal. El paciente puede además presentar cefalea o pérdida de la audición y estenosis espinal lumbar u otros síndromes de compresión nerviosa con déficits sensoriales o motor. Los síntomas por afectación craneal corresponden a deformidad craneal, pérdida de audición por afectación coclear, cefalea, mareos, vértigo y raramente hidrocefalia con inestabilidad para la marcha, demencia, apatía por robo sanguíneo por la alta vascularización craneal. Puede existir impresión basilar por afectación de la base craneal. Es posible ver venas en el cuello cabelludo dilatadas.
La enfermedad se diagnostica principalmente mediante el examen radiológico. temprano en el curso de la enfermedad, predomina la actividad lítica, causando lesiones osteolíticas focales generalmente en las áreas frontales y occipitales. Posteriormente, las áreas de la esclerosis desarrollan, inicialmente a predominio de la tabla interna más que la externa que se adelgaza por el aumento del diploe, las suturas no son barrera para la enfermedad, lo que conduce a los aspectos característicos de lítico mixtos y áreas escleróticas, con trabéculas engrosadas, expansión ósea, engrosamiento cortical y deformidad. El diagnóstico diferencial se debe establecer con las metástasis escleróticas o líticas deben tenerse en cuenta por ello una exploración ósea con radioisótopo puede ser recomendada en todos los pacientes como parte de la evaluación de diagnóstico inicial para determinar la distribución de la enfermedad, en particular, la participación de los sitios con el potencial de complicaciones, tales como la base del cráneo, la columna vertebral y los huesos largos . La tomografía computarizada es útil para evaluar la participación de la base del cráneo, especialmente para los pacientes con sordera, la estenosis espinal u otras complicaciones neurológicas. En las radiografías de cráneo, se observa osteolisis avanzada como grandes áreas de radiolucidez, generalmente en los huesos frontal y occipital y se designa como “osteoporosis circunscrita”. Posteriormente el marcado engrosamiento de la tabla interna de la calota produce importante aumento del espacio diploico conocido como un cráneo “Tam O’shanter” . El aspecto clásico de “algodón” es causado por áreas irregulares de osteosclerosis focal como se ha visto en el presente caso.
En el diagnostico diferencial de las lesiones craneales osteolíticas deben tomarse en cuenta diferentes etiologías: 1.- Congénitas y del Desarrollo, 2.- Traumáticas, 3.- Inflamatorias, 4.- Neoplasias, 5.- Misceláneas. En el grupo de las enfermedades congénitas se encuentran los quistes epidermoides con su borde esclerótico que es característico. La segunda lesión de este grupo seria la displasia fibrosa que es una condición benigna donde el hueso es substituido por tejido fibroso con frecuencia ocurre en el frontal, base craneal, huesos faciales. A diferencia del Paget la edad de aparición más frecuente es adolecentes y adultos jóvenes. En la tomografía de cráneo la imagen de vidrio esmerilado es característica de la displasia fibrosa que histológicamente corresponde a pequeñas calcificaciones sobre un estroma fibroso, Existen 3 variedades de esta lesión: A.- Quística caracterizada por engrosamiento del diploe adelgazamiento de la tabla externa y poca afectación de la tabla interna. B.- Esclerótica que afecta generalmente la base especialmente el esfenoides y los huesos faciales.
C.- Mixta donde coinciden las lesiones osteolíticas con las lesiones escleróticas. Algunos de los signos radiológicos que permiten su diferenciación son: imagen vidrio esmerilado en la tomografía computada, la simetría de las lesiones es más frecuente en Paget mientras que la afectación de maxilar, esfenoides, orbita y cavidad nasal.
Los hallazgos de laboratorio generalmente muestran aumento de la fosfatasa alcalina, generalmente es proporcional el aumento al grado de actividad de la enfermedad, pero se pueden encontrar valores normales o ligeramente aumentados en enfermedad monostótica o poliostótica. Se pueden encontrar exámenes normales en lesiones pélvicas aisladas o cuerpos vertebrales afectados o en enfermedades donde predomine la etapa esclerótica en contraste se puede observar valores muy elevados en lesiones craneales .
Existen otros marcadores de laboratorio como son el procolagen tipo I, N-propeptido (PINP), C-telopeptide (CTx), N-Telopeptido urinario (NTx), Hidroxiprolina urinaria aunque se han descrito paciente con gammagrafía que demuestra actividad de la enfermedad con valores de estas pruebas normales .
Cuando las características clínicas, de laboratorio y radiológicas son características de la enfermedad de Paget, no es necesaria la realización de la biopsia, pero cuando los diagnósticos de enfermedad metastásicas como carcinoma prostático, mieloma múltiple, etc., y estos no están claros se puede realizar. La otra indicación de biopsia es cuando esta enfermedad se presenta en áreas de poca frecuencia, como en este caso.
CONCLUSIONES
La enfermedad de Paget es una patología que se presenta después de los 50 años, existen áreas geográficas de alta incidencia Inglaterra, Escocia, Europa central. Se caracteriza por un alta tasa de recambio del hueso donde factores genéticos y ambientales están involucrados, múltiples locis genéticos han sido identificados. La mayoría de los pacientes con esta enfermedad son asintomáticos, el cráneo es uno de los sitios afectados con frecuencia, manifestando por cefalea, deformidad craneal, hipoacusia. Las pruebas de laboratorio muestran el aumento del recambio óseo, las lesiones osteolíticas se ven en las etapas tempranas, mientras que en las avanzadas se observa el aumento de grosor y crecimiento óseo. En la mayoría de estos paciente la fosfatasa alcalina esta elevada y otras pruebas de laboratorio no son necesarias, la biopsia está indicada cuando existan dudas en el diagnóstico.
REFERENCIAS
- Paget SJ. On a form of chronic inflammation of bones (Osteitis deformans). Medical Chirulogical Transaction 29-53(1877).
- Van Staa TP, Selby P. Incidence and natural history of Paget disease of bone. England and Wales J. Bone Miner. Res. 17, 465-471 (2002).
- Dunphy, L.M., Winland-Brown J.E.. Primary care. The art and Science of advanced practice nursing Philadelphia 2001.
- Altman RD, Bloch DA, Hochberg MC, Murphy WA. Prevalence of pelvic Paget’s disease of bone in the United States. J Bone Miner Res. 2000; 15:461.
- Barker D.J, Chamberlain A.T. Paget´s disease of bone. The Lancashire focus. BMJ 280, 1105-1107 (1980).
- Barker D.J. The epidemiology of Paget’s disease. Metab Bone Dis Relat Res 1981; 3:231.
- Paget’s Disease: A Case in Point Nivens, Anita S Orthopaedic Nursing; Nov/Dec 2004; 23, 6.
- Sutton, D. (ed.) (1975) A textbook of Radiology, 2nd edn, p. 78. Churchill Livingstone, Edinburgh.
- Cremers S, Garnero P. Biochemical markers of bone turnover in the clinical developement of drugs for osteoporosis and metastasic bone disease; potencial uses and pitfalls. Drugs 66 , 2031-2058 (2006).
- Alvares L, Ricos C, Peris P. Components of biological variation of biochemical markers of bone turnover in Pget´s bone disease. Bone 26, 571-576 (2000).
- Price C.H. The incidence of osteogenic sarcoma in south West England an its relationship to Paget´s disease of bone. J. Bone Joint Surg Br. 44-B, 366-376, 1962.
- Seton M., Choi H.K., Hasen M.F. Analysis of enviromental factors in familial versus sporadic Paget´s disease of bone. The New England Registry for Paget´s Disease of Bone. J. Bone Miner. Res. 18,1519-1524.2003.
- Seton M. Paget´s disease; epidemiology and pathophysiology. Current. Osteopros Rep 6, 125-129 (2008).
- Bastin S, Bird H, Gamble G, Cundy T. Paget’s disease of bone: Becoming a rarity?. Rheumatology (Oxford) 2009;48:1232–5.
- Bae KB, Kwon JH, Kim YH, Jung TY, Cho JH. Juvenile Paget’s disease with paranasal sinus aplasia. Clin Exp Otorhinolaryngol. 2008;1:224–6.
- Karunakaran K, Murugesan P, Rajeshwar G, Babu S. Paget’s disease of the mandible. J Oral Maxillofac Pathol. 2012;16:107–9.
- Chung PY, Beyenes G The majority of the genetic risk for Paget´s diseaseof bone is explained by geneticvariants closet o de CSF1, OPTN,TM7SF4 and TNFRSF11 genes. Hum Genet 2010;128:615
- Laurin N, Brown JP Recurrent mutation of the gene encode sequestosome 1 (SQSTM1/P62) in Paget disease of bone.Am J Hum Genet 2002;70:1582
- Lucas GJ,Riches PL Hocking LJ Identification of a mayor locus for Paget disease on chromosome 10p13 in families of British descent.J Bone Miner Res 200;23:58
- Devogelear JP, Bergmann P, Body JJ et al. Management of patient with Paget disease: a consensus document of Belgian Bone Club. Osteoporos Int 2008;19:1109
- Butt ST, Fatima S, Butt R, Nasir W, Jameel G, Irfan J. Polyostotic Paget’s disease. J Coll Physicians Surg Pak.2012;22:461–3.
- Bhargava P, Maki JH. Images in clinical medicine. “Cotton wool” appearance of Paget’s disease. N Engl J Med. 2010;363:e9.
- Seton M Paget´s disease of bone in: Rheumatology 4th Hochberg MC, Silman AJ Elservier, Philadelphia 2008. P.2003
- Harink HI, Bijvoet OL, Blanksma HEfficacious management with aminobiphosphonate in Paget´s disease of bone. Clin Orthop elat Res 1987;79.
